Abdelaziz A. Mohamed1*, Manal M. Saad 1, Sherif H. Abdeen 2, Mona K. Marei 1
1Tissue Engineering Laboratories, Faculty of Dentistry, Alexandria University, Egypt. 2Department of Zoology, Faculty of Science, Mansoura University, Egypt.
*Corresponding Author: Dr. Abdelaziz Mohamed. Email: [email protected]
DOI: http://dx.doi.org/10.14206/canad.j.clin.nutr.2013.01.06
Abstract
Background: The utilization of stem cell trans-differentiation into insulin-producing cells (IPCs) would provide potential promising therapy for diabetes mellitus (DM). Objective: The study was aimed to investigate the differentiation potential of rabbit’s bone marrow-derived cells into insulin producing cells. Methods: Bone marrow-derived mesenchymal stem cells (MSCs) were obtained from three New-Zealand male white rabbits and propagated in a primary culture for 14 days in low glucose Dulbecco’s Modified Eagle’s Medium (DMEM), harvested and subjected to another three passages encompassing 21 days. After that, MSCs were functionally defined by their ability to differentiate into osteoblasts. This was achieved by incubation with the DMEM containing 10-7 M dexamethasone, 10 mM β-glycerophosphate and 50 µl/ml of ascorbic acid. Osteogenic differentiation was followed up at days 2, 4, 7 and 9 by staining cells with alizarin red S and vonKossa. Results: It was found that the onset of differentiation was at day 7 and continued at day 9. On the other hand, another patch of MSCs were induced into insulin-producing cells by two step incubation. The first with high glucose serum-free DMEM containing 0.5 mmol/L β-mercaptoethanol for three days and the second follows by incubation with the same medium containing 10 mmol/L nicotinamideinstead of β-mercaptoethanolfor 18 days. Trans-differentiation was followed up at days 4, 9, 14 and 21. It was found that the cells have trans-differentiated into insulin-producing cells starting from day 14 and continued in the subsequent days as judged by their affinity to stain with diphenylthiocarbazone (dithizone or DTZ) at days 14 and 21. Conclusion: The results would provide some insights of using rabbit’s bone marrow as a source of MSCs with their differentiation potentials. This might help for the development of a future stem cell therapy for diabetes as well as several other human diseases.
Full Text
INTRODUCTION
Stem cells are undifferentiated cells which have the ability to renew themselves through mitotic cell division and differentiate into a diverse range of specialized cell types (1). Of these cells, mesenchymal stem cells (MSCs) have a large capacity for self-renewal while maintaining their multipotency. MSCs display multiline age differentiation potentials into a variety of cells such as osteoblasts, adipocytes and chondrocytes as well as myocytes and possibly neuron-like cells (2). They are capable of suppressing immune responses and offer therapeutic potential for achieving transplantation tolerance (3). Accordingly, MSCs have been considered as an appropriate source for cell and gene therapy tools for treatment in a number of injuries, congenital and degenerative diseases including; spinal cord injury (4), osteogenesis imperfecta (5), stroke (6), parkinsonism (7) and diabetes mellitus (8).
Diabetes mellitus refers to a group of chronic metabolic diseases characterized by hyperglycemia due to defects in insulin secretion, insulin action, or both (9).Type I, insulin-dependent, diabetes is the result of autoimmune destruction of insulin-producing pancreatic β-cells. On the other hand, in type two or non-insulin-dependent diabetes, insulin production is inadequate mainly because of the peripheral insulin resistance and subsequent β-cell apoptosis. Diabetes and its devastating effects, which include retinopathy, nephropathy, stroke and heart attack, afflict more than 194 million people worldwide. According to the World Health Organization, these numbers will be more than double by 2030 (10, 11).
Survival of patients with insulin-dependent diabetes relies on recurring insulin delivery, which does not cure the disease or prevent diabetes-associated maladies. Although, Type 2 diabetes can be managed through a combination of diet, exercise and prescription of drugs, almost 30% of the affected people will require frequent administration of insulin. Therefore, the development of therapies to replace insulin regimens is highly desirable. To that end, islet transplantation has afforded promising results, with some patients experiencing insulin independence for more than 5 years after the initial procedure (12). Ongoing procedural improvements are underway to increase the time span of liberation from insulin and reducing the side effects due to immunosuppressant. However, the scarcity of available donor tissues hinders wide application of pancreas/islet transplantation. Thus, the hope stands in finding renewable sources of islet β-cells such as pancreatic β-cell lines, embryonic stem cells (ESCs), adult progenitor cells (APCs), regenerating native islet cells (13) and MSCs (14).
From all of the previously mentioned literatures, it was therefore of interest to carry out experiments investigating the differentiation potential of rabbit’s bone marrow-derived stem cells into insulin-producing cells as a first step for their future use in islet transplantation purposes.
Materials and methods
Abbreviations
The following abbreviations are used; Adult progenitor cells (APCs), deionized distilled water (ddH2O), diabetes mellitus (DM), dimethyl sulfoxide (DMSO), diphenylthiocarbazone (dithizone or DTZ), Dulbecco’s Modified Eagle’s Medium (DMEM), embryonic stem cells (ESCs), fetal bovine serum (FBS), hours (hr), insulin I and II, glucose transporter-2 (Glut-2), insulin-producing cells (IPCs), intra-muscular (IM), mesenchymal stem cells (MSCs), minutes (min), mononuclear cells (MNCs), pancreatic duodenal homeobox-1 (Pdx-1) and phosphate buffer saline (PBS).
Animal work
All animal experiments were performed according to the guidelines of the National Institutes of Health (NIH) for laboratory animal use (15). All procedures applied in this study were of no pain category (category C).
Aspiration bone marrow-derived cells
Bone marrow aspiration was performed according to the method of Horan et al. (16).Three male New-Zealand white rabbits (obtained from the research farm, Faculty of Agriculture, Alexandria University) were anesthetized by intra-muscular (IM) injection with 5 mg/kg xylazine as a preanaesthetic medication. After 5 min, animals were intra-muscularly injected with 30 mg/kg ketamine-HCl. All surgical procedures were performed in a biological safety cabinet (Nuaire biological safety class II, T-Telestar, Spain) with surgery room disinfected overnight by UV light. The fur of the thigh was shaved and skin disinfected. Skin incision was made to expose the rabbit’s femur. A hole was drilled in the femur using a surgical rose-head bur. A sterile plastic aspiration needle containing 8 µl heparin/ml sample was used. The marrow sample was kept in a sterile package till transferred to the cell culture laboratory. The surgical wound was sutured and the rabbit was taken back to the animal care unit. All animals tolerated the surgical procedures for bone marrow aspiration. No significant changes in body weight, no postoperative infections were observed.
Cultivation media
All buffers, media and their supplements were purchased from BioWhittkers® Lonza, Verviers, Belgium unless otherwise noted. These media included: Basal medium
[Low glucose Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 mmol glucose/L and supplemented with10% (v/v) fetal bovine serum (FBS),1% L-glutamine,10 IU/ml penicillin/streptomycin and 20 mM HEPES]. Ostogenic differentiation medium [Low glucose DMEM with4.5 mmol glucose/L and supplemented with 10% FBS, 1% L-glutamine, 1% penicillin/streptomycin, 20 mM HEPES,10 mM β-glycerophosphate(Sigma, USA),50 µg/ml ascorbic acid (Sigma, USA) and10-7M dexamethasone (Sigma, USA)].
First step trans-differentiation medium: Serum-free high glucose DMEM with 25 mmol glucose/L and supplemented with 20 mM HEPES,1% L-glutamine,1% penicillin/streptomycin and 0.5 mmol/L β-mercaptoethanol (Sigma Aldrich Co., Germany). Second step trans-differentiation medium: Serum-freehigh glucose DMEM with 25 mmol glucose/L and supplemented with 20 mM HEPES buffer,1% L-glutamine,1% penicillin/streptomycin and10 mmol/L nicotinamide (Sigma Aldrich Co., Germany).
Media preparation
The powder supplements of the media were dissolved in deionized distilled water (ddH2O) obtained from Barnstead water deionization system (Barnstead-Thermolyne, USA). In case of using another dissolvent, it was sterilized by membrane filtration through 0.22 μm microbiological filter system (TPP-Europe, Switzerland). After adding the supplements, the pH of the medium was adjusted at 7 and medium was then sterilized by membrane filtration. A sterility test was performed for 3 days at 37°C. Medium contamination was detected depending on the color change of the phenol red, which gives a yellow-orange color through the change in pH to acidic value as a function of the microbial growth. A sample was also examined microscopically after passing the sterility test. All media were stored at 4°C until being used.
Isolation of rabbit’s bone marrow-derived mesenchymal stem cells
The method of Sun et al., (17) was used with minor modifications. Bone marrow samples of about 1.5 to 2 ml were aspirated from each rabbit and transferred to the cell culture laboratory in a sterile package. Inside a biological safety cabinet class II (Thermo electron corporation, Germany) samples were diluted with a double volume of the basal medium, collected by centrifugation at 390 g for 7 min at room temperature and washed twice with phosphate buffer saline, pH 7.4 (0.0067 M PBS without calcium and magnesium).
The samples which have clots or bone marrow tissue were filtered through 200 µm filter (Sigma Aldrich Co., Germany) using PBS for washing and liberation of the trapped cells in the tissue. Bone marrow mononuclear cells (MNCs) were then collected by centrifugation, washed twice with the basal medium and resuspended in one ml of the same basal medium, counted (10 ml of cells + 10 µl of 0.4% methyl violet + 180 µl glacial acetic acid) and tested for viability using hemocytometer. MNCs were then seeded in polystyrene coated T-flasks at a density of 5.5 x 106cells/cm2and incubated at 37oC, 5% Co2 and 95% humidity. Except for the first four days which were required for cell adhesion, the cells were fed twice a week by washing with pre-wormed PBS then adding fresh medium. The cells were allowed to proliferate till reaching the 100% confluence in 14 days (the primary culture).
Only the cells which attach to the polystyrene surface were allowed to grow while suspended ones were discarded during washing. Cell morphology was followed up daily using a phase contrast inverted microscope (NIKON-Japan) using (SIS) software system for capturing and analysis of images. All cultures were independently repeated for three times.
Confluent monolayer of cells were harvested by trypsinization using trypsin/EDTA buffered solution 6 ml of 0.5% trypsin, 200 mg/L versene EDTA (Gibco/Invitrogen, Germany) at 37oC for 10 min. The suspended cells were rapidly neutralized with a double volume of the basal medium to stop the action of trypsin. The cells were then collected by centrifugation, washed twice with pre-wormed PBS, counted and tested for viability. The cells were then reseeded in fresh medium at a density of 1400 cells per cm2 in a new cell culture T-flasks. The cells were allowed to grow for three subsequent passages of 7 days each. After passage three, MSCs were subjected to osteogenic differentiation and trans-differentiation into insulin-producing cells (18).
Estimation of viable cell concentration
Viable cell concentration was determined using trypan blue exclusion method (19). Ten ml of cells were diluted to 1:2 in 0.4% trypan blue stain. The hydrophilic trypan blue diffuses through cell membrane of the dead cells. Hence, the viable cells appear bright while the unviable ones appear blue. Cell viability was determined according to the following equation: Cell viability = Total number of viable cells/Total cell count ×100
Cryopreservation of bone marrow-derived cells
At the end of each culture, a separate patch of cells were trypsinized and collected for cryopreservation (20). The cells were cryopreserved at a density of 1×106 cells/ml in freezing medium; basal medium with 25% FBS and 5% v/v dimethyl sulfoxide (DMSO). The cells were then stored at -156ºC in cryo-vials (Nunc, Wiesbaden, Germany) in the vapor phase of a liquid nitrogen locator (Cryo Biological Storage System Locator 4 plus thermolyne, USA).After several pilot trials, cryopreservation of cells was optimized in 5% DMSO as a cryoprotectant. This protocol yielded the highest cell viability of 69% upon revitalization. In order to revitalize the cells, cryogenic-vials were warmed in a water bath at 37oC for 2 min and resuspended rapidly in FBS, centrifuged for 5 min, washed twice with PBS, counted, tested for viability and resuspended in the basal medium. Counting and testing viability were done before and after cryopreservation as a routine procedure.
Characterization of rabbit’s bone marrow-derived MSCs
Rabbit’s bone marrow-derived MSCs were characterized by their induction into osteogenic differentiation using the method of Nadriet al.,(21). The cells were incubated with the osteogenic differentiation medium at the end of passage three, especially, when they have reached 80-90% confluency. The cultures, in 6-well plates, last for two weeks with cells fed twice a week. Cell morphology was followed up daily. To detect the onset of osteogenic differentiation, cells were stained with alizarin red S and von Kossa at days 2, 4, 7 and 9.
Alizarin red S staining of the differentiated osteoblasts
Calcium depositions of osteoblasts were detected through its reaction with alizarin sulphate with scarlet to red color indicating positive results (22). For staining, the cells were washed with pre-warmed PBS, fixed in 4% formaldehyde for 30 min. The fixative was then removed and the cells were washed using distilled water. The alizarin red S stain pH 4, (2% alizarinsulphonate; LobachemieCo., India) was added to cover all the cells for approximately 5 min at room temperature. The stain was then discarded and the cells were washed again with distilled water to remove the excess of stain then air dried. Red patches in the tested cultures were examined microscopically. Cells propagated in the basal medium were included as control cultures.
vonKossa staining of the differentiated osteoblasts
vonKossa staining was applied to confirm the osteogenic differentiation of MSCs (23). Like the staining protocol of alizarin red S, the cells were washed with pre-warmed PBS, fixed for with 4% formaldehyde 30 min and washed with distilled water. The cells were then incubated with 1% aqueous silver nitrate solution (Fisher scientific-UK) in a clear glass coplin jar placed under a 60-100 watt light bulb for 1 hr. After that, the cells were carefully washed with distilled water to eliminate all the debris of silver nitrate and incubated with 5% sodium thiosulfate for 5 min. The solution was discarded and the cells were washed again with distilled water and air dried. The progress of the reaction was followed up visually. The staining intensity of the brown to black color was a reference for the presence of calcium produced by the differentiated osteoblasts.
Trans-differentiation of MSCs into insulin-producing cells
At the end of passage three, especially when MSCs have reached 80-90% confluence, the cells were induced into insulin producing cells through two steps. The first step included the incubation of the cells with the 1ststep trans-differentiation medium for three days. This was followed by a careful wash with pre-warmed PBS and incubation with the 2ndstep trans-differentiation medium for 18 days. All over the two steps, the cells were fed twice a week (17).
Staining of insulin-producing cells with dithizone
Dithizone (DTZ), a zinc-chelating agent, is known to selectively stain pancreatic β cells by crimson red color. In the secretory granule of a mature insulin-producing cell, every 6 insulin molecules are coordinated by a Zn atom giving the red color. For staining, the cells were incubated with DTZ stain (Lobachemie Co., India; 10 mg/ml DTZ in DMSO diluted to 1:100 in the basal medium) for 30 min. The cells were then carefully washed with pre-wormed PBS for three times. The staining result was monitored under an inverted microscope. After 5 hours, the stain was completely disappeared and the cells were able to grow successfully (24).
Data analysis
Statistical analyses were performed with one-way ANOVA followed by SPSS software version 10 (SPSS Science, Chicago, IL, USA) analysis. Data (mean ± SD) were considered statistically significant at a value of P < 0.05. ANOVAtest was applied to compare the growth kinetics during the different time points of the expansion period i.e. the primary culture and the first two passages.
Results
Isolation of bone marrow-derived mesenchymal stem cells
Isolation of bone marrow-derived MSCs required a primary culture of 14 days and three subsequent passages of 21 days. This can be summarized as follows; bone marrow samples of 1.5 – 2 ml were processed for isolation of mononuclear cells (MNCs) giving rise to the mean numbers of cells 3.9×107. MNCs were then seeded in a primary culture at a density of 5.5×105 cells/cm2 giving a yield of 15.6×103 cells/cm2 at the end the primary culture (day 14). Whereas, the number of cells seeded in passage one and two was 1.4×103 cells/cm2 and the number of cells yielded was 8.4×103 and 7.2×103cells/cm2 for passages one and two, respectively. The cells were allowed to grow in passage three similar to that of passages one and two with one difference of being differentiated or trans-differentiated at the 100% confluency of passage three without transfer to a next passage (Table 1). Except for day zero of the primary culture, comparison of the growth kinetics of all culture during the expansion period (the first 5 weeks) revealed that all cultures were significantly grow (P < 0.05) starting from day 2 after reseeding.
In the primary culture, MSCs were allowed to adhere to the polystyrene tissue culture flask surface in the first four days without media change. Whereas, at day five all of the co-cultured red blood corpuscles (RBCs) and cell debris were discarded with the washing step prior to the media change. At day five, MSCs showed an anchorage-dependent growth with their attachment confirmed via the emergence of expanded cytoskeleton processes. In the following days, cells proliferate with increased cell to cell contact giving rise to cell colonies. By the end of the primary culture, at day 14, the cell proliferation reached about 90% confluence. Thus, the cells were trypsinized and reseeded in another culture (Figure 1).
In passage one and unlike the primary culture, the cells adhere in few hours and overnight incubation was sufficient to make the cells to completely adhere in day 1. However in day 2, the cells started to contact with each other. In the subsequent days, the cells proliferated, forming colonies and their morphology changed to the spindle-shape. The cells reached the 100% confluence in only 7 days (Figure 2). After that, the cells were collected by trypsinization and reseeded in a new cell culture flask for another passage. The number of cells at the end of passage one was 8.2×103 cells/cm2 with doubling time of 21.6 hr. The viability after harvesting was 98%.
However in passage two, the cells showed similar manner of cell growth. They showed substratum adherence, migration, cell-cell contact, colony formation and confluence. The cell yield of passage two was 7.2×103 cells/cm2 and doubling time was 17.63 hr.
Characterization of MSCs by osteogenic differentiation
Cells were incubated in conditioned medium of dexamethasone, β-glycerophosphate and ascorbic acid. They were following up daily with an inverted microscope. The cells were stained with alizarin red S and von Kossa stains in days 2, 4, 7 and 9 to determine the onset of osteogenic differentiation. All tested cultures showed positive results starting from day 7. However, controls showed negative staining all over the four time points.
On the other hand, the morphology of the cells under the stress of osteogenic differentiation showed the spindle-shape in the tested cultures. The controls showed some aspect of over confluent appearance. The difference in cell morphology between the tested and control cultures during the first few days till day 7 was not clear but with staining the discrimination was possible (Figure 3).
Trans-differentiation of MSCs into insulin-producing cells
MSCs were induced into insulin-producing cells via two steps. The first step included the incubation of cells with high glucose serum-free DMEM containing β-mercaptoethanol. Upon trans-differentiation, the cells showed aggregated colonies. The first step of differentiation was extended to 3 days then the second step follows at day 4 by replacing β-mercaptoethanol with nicotinamide. At day 4, the colonies were formed and developed in the subsequent days. At day 14, the morphology of cells was changed to the rounded to oval shape and continued with this morphology in days 19 and 21 (Figure 4).
The dithizone (DTZ) staining was applied to test the production of insulin at different time points including days 4, 9, 14 and 21. While, the tested cultures showed an affinity to the DTZ stain (crimson red color) starting from day 14 and continued in day 21, the controls showed negative results all over the time of the experiment (Figure 5).
Discussion
Mesenchymal stem cells are multipotentstem cells that can differentiate into a variety of cell types (25). Bone marrow-derived MSCs, also named colony-forming fibroblastic cells (26), marrow stromal stem cells (27) and mesenchymal progenitor cells (17), have been isolated and characterized from many species including: rats, cats, dogs, baboons and rhesus monkeys, rabbits, pigs, goats and sheeps (21). MSCs have many advantages including: the ease of isolation, the extensive multi-lineage differentiation potential (18, 28), the hypo-immunogenic property which modulate the lymphocytic functions, the possibility of allogenic transplantation, the capability of systemic transplantation for generalized diseases as well as the local implantation for the local tissue defects. MSCs can also be used as a vehicle for genes in gene therapy protocols or to generate transplantable tissues and organs in tissue engineering protocols. Accordingly, MSCs are among the first stem cell types to be introduced in the clinic and several encouraging clinical trials are under way to study the efficacy and long-term safety of therapeutics based on MSCs (29, 30).
Several recent studies have demonstrated the feasibility of generating insulin-producing cells obtained from progenitor cells of various cellular sources, including the pancreas, liver and intestinal epithelium, as well as, the pluripotent embryonic stem cells of mouse and human origin. However, even with the conceptual advances offered by these findings, some obstacles, such as the immune rejection and autoimmunity against newly formed cells derived from pancreatic stem cells, still remain. Despite their promising potential, it may also prove difficult to obtain enough autologous adult stem cells from these organs. To overcome these limitations, the possibility of using rabbit bone marrow derived cells was explored as a source of insulin-producing cells under specific in vitro culture conditions. Bone marrow has been known for years to represent a safe and abundant source for large quantities of adult stem cells (13).
In the present study, the source of MSCs was the bone marrow of New-Zealand white rabbits. This animal model was chosen due its availability, the ease of handling and the ease of choice between in and out-bred. In case of aspiration of bone marrow samples from mice or rats, the animal is often killed before aspiration. Contrarily, there is no need to kill the rabbit to aspirate a bone marrow sample with the advantage of repeating aspiration within 8 weeks (16).
Several techniques have been developed to obtain pure cultures of MSCs by the reduction or elimination of non-MSCs from bone marrow cultures (31). This was achieved, in the present study, by the application of a prolonged expansion step through which all of the bone marrow-derived cells pass in a primary culture for 14 days and three subsequent passages of 21 days. During this time, only MSCs, due to their adherent property, were retained in the culture while all of the other non-adherent cells were eliminated by washing during media change. Moreover, the selective medium and the polystyrene-coated tissue culture flasks enhanced the adhesion properties of the MSCs (21).
Cultures of MSCs is usually contaminated by different types of non-MSCs adherent cells like monocytes and macrophage (32) and as MSCs are more responsive to trypsin, they were purified in the present study with an optimized concentration of trypsin/EDTA (17). Unlike the primary culture when the cells needed 14 day to reach the 100% confluency, the behavior of the cells changed in the subsequent passages as they reached the 100% confluency in only 7 days. This could be owing to the gradual selection of a learned mesenchymal cell clones with proceeding passages (33).
One of the hallmarks of MSCs is their multi-potency, defined as the ability to differentiate into several mesenchymal lineages including; bone, cartilage, tendon, muscle, marrow stroma and adipose tissue. Usually, the tri-lineage differentiation into bone, adipose tissue and cartilage is taken as a criterion for the multi-potentiality of these cells (34). In the current study, MSCs were differentiated into osteoblasts using a conditioned medium containing dexametasone, ascorbic acid and β-glycerophosphate. The differentiation was followed up at different time points including days 2, 4, 7 and 9 through staining with alizarin red S and von Kossa stains. The onset of osteogenic differentiation was at day 7 as judged by the positive staining affinity of the calcium depositions of the osteoblasts with the two stains. These results confirmed the mesenchymal stem lineage of the processed cells
(35).
Upon trans-differentiation, the presence of insulin was evidenced using the dithizone stain (DTZ) which is a zinc chelating agent. It does not require prior fixation and stains insulin molecules either in the cells or in the media.DTZ disappears after 5 hr leaving live cells able to complete their life cycle (24, 36). The onset of trans-differentiation was followed up at different time points including; days 4, 9, 14 and 21. It was found that a complete trans-differentiation was obvious in day 21 while partial trans-differentiation was observed in day 14. In contrast, there were no signs of trans-differentiation at days 4 and 9. Despite DTZ is a vital stain, the staining intervals were chosen minimal to decrease the stress applied on the cells at every staining time.
On the other hand, the microscopic examination for the trans-differentiation revealed aggregations like clusters of insulin producing cells with a diameter ranging from 100 to 150µm. These clusters appeared from day 2 of differentiation and continued to develop till day 12 where the cells acquired the spindle-shape. This morphology was changed to the rounded and oval shapes at day 14 which may be considered as a sign for the meso-endodermal differentiation (37).
There are two key steps in culture conditions that appear important for inducing the trans-differentiation of MSCs cells into insulin-producing islet-like cells. First, the transfer of cells from an expansion period in low-glucose medium to differentiation in high-glucose medium until certain genes, such as pancreatic duodenal homeobox-1 (Pdx-1), insulin I and II, glucose transporter-2 (Glut-2), and islet amyloid polypeptide, become detectable. Second, in order for the MSCs cells to become glucose responsive, further differentiation and maturation are required through either in vitro culture with cell promoting factors, such as nicotinamide, mercaptoethanol or transplantation of the cells into diabetic animals (17, 38). It is well known that glucose is a growth factor for cells. It promotes cell replication in vitro and in vivo and increases insulin content of the cell (39).
Nicotinamide is a poly (ADP-ribose) synthetase inhibitor known to differentiate and increase cell mass in the cultured human fetal pancreatic cells (40). It protects the cells from desensitization induced by the prolonged exposure to large amounts of glucose. It have also been demonstrated that nicotinamide promoted the formation of fetal porcine islet-like cell clusters and increased the rates of pro-insulin biosynthesis in these clusters. Moreover, the stimulatory effects of nicotinamide on insulin production and content by fetal porcine islet-like cell clusters result from neo-formation of cells through differentiation (41). Another study described how nicotinamide-treated islets derived from the pancreatic progenitor cell had more insulin and secreted significantly more insulin than cultures treated with glucose alone (42).
The present study used the low glucose medium for cell expansion and for induction to insulin-producing cells the medium was changed to high glucose and this was the first key step in trans-differentiation. The second key step was achieved through culturing with the media containing β-mecaptoethanol then nicotinamide as cell promoting factors.
In conclusion, the present data suggest potential uses of MSCs from rabbit’s bone marrow in induction into insulin-producing cells. This provides hope for a future development a stem cell therapy for diabetes as well as several other human diseases and injuries.
References
- Becker AJ, McCulloch EA, Till JE. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963; 197:452–454.
- Engler AJ, Sen S, Sweeny HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006; 126(4):677–689.
- Ding Y, Bushell A, Wood KJ. Mesenchymal stem-cell immunosuppressive capabilities: therapeutic implications in islet transplantation. Transplantation. 2010; 89(3):270-273.
- Awad HA, Butler DL, Boivin GP, Smith FN, Malaviya P, Huibregtse B, Caplan AI. Autologous mesenchymal stem cell-mediated repair of tendon. Tissue Eng. 1999; 5(3):267-277.
- Bancroft JD, Gamble M. Theory and practice of histlogical techniques, Fifth Edition, 2002, pp: 243-267.
- Bonner-Weir S, Sharma A. Are there pancreatic progenitor cells from which new islets form after birth? Nat Clin Pract Endocrinol Metab. 2006; 2(5):240-241.
- Chen J, Zhang ZG, Li Y, Wang L, Xu YX, Gautam SC, Lu M, Zhu Z, Chopp M. Intravenous administration of human bone marrow stromal cells induces angiogenesis in the ischemic boundary zone after stroke in rats. Circ Res. 2003; 92(6):692-699.
- Digirolamo CM, Stokes D, Colter D, Phinney DG, Class R, Prockop DJ. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol. 1999; 107(2):275-281.
- Eslaminejad MB, Nikmahzar A, Taghiyar L, Nadri S, Massumi M. Murine mesenchymal stem cells isolated by low density primary culture system. Dev Growth Differ. 2006; 48(6):361-370.
- Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV.Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966; 16(3):381-390.
- Harris MI. Newly revised classification and diagnostic criteria for diabetes mellitus. In: Taylor ST, ed. Current Review of Diabetes. Philadelphia: Current Medicine, Inc. 1999, pp: 1-9.
- Hogan P, Dall T, Nikolov P; American Diabetes Association. Economic costs of diabetes in the US in 2002. Diabetes Care. 2003 ; 26(3):917-932.
- Horan PK, Muirhead KA, Gorton S, Irons RD. Aseptic aspiration of rabbit bone marrow and enrichment for cycling cells. Lab Anim Sci. 1980; 30(1):76-79.
- Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, Muul L, Hofmann T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesisimperfecta: Implications for cell therapy of bone. Proc Natl Acad Sci USA. 2002; 99(13):8932-8937.
- Hurwitz DR, Kirchgesser M, Merrill W, Galanopoulos T, McGrath CA, Emami S, Hansen M, Cherington V, Appel JM, Bizinkauskas CB, Brackmann HH, Levine PH, Greenberger JS. Systemic delivery of human growth hormone or human factor IX in dogs by reintroduced genetically modified autologous bone marrow stromal cells. Hum Gene Ther. 1997; 8(2):137-156.
- In ‘t Anker PS, Scherjon SA, Kleijburg-van der Keur C, de Groot-Swings GM, Claas FH, Fibbe WE, Kanhai HH. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta.Stem Cells. 2004; 22(7):1338-1345.
- Inada M, Follenzi A, Cheng K, Surana M, Joseph B, Benten D, Bandi S, Qian H, Gupta S. Phenotype reversion in fetal human liver epithelial cells identifies the role of an intermediate meso-endodermal stage before hepatic maturation. J Cell Sci. 2008; 121(7):1002-1013.
- Krupnick AS, Shaaban A, Radu A, Flake AW.Bone marrow tissue engineering. Tissue Eng. 2002; 8(1):145-155.
- Latif ZA, Noel J, Alejandro R. A simple method of staining fresh and cultured islets. Transplantation. 1988; 45(4):827-830.
- Lee MW, Cho, J, Yang MS, Moon YJ, Park JS, Kim HC, Kim YJ. Mesenchymal stem cells from cryopreserved human umbilical cord blood. Biochem Biophys Res Commun. 2004; 320:273-278.
- Lock LT, Tzanakakis ES. Stem/Progenitor cell sources of insulin-producing cells for the treatment of diabetes. Tissue Eng. 2007; 13(7):1399-1412.
- Menon LG, Shi VJ, Carroll RS. Mesenchymal stromal cells as a drug delivery system. In: Melton D, Girard L, editors. Stem Book Cambridge (MA): Harvard Stem Cell Institute; 2008-2009.
- Mishra PK, Singh SR, Joshua IG, Tyagi SC. Stem cells as a therapeutic target for diabetes. Front Biosci. 2010; 15:461-477.
- Nadri S, Soleimani M, Hosseni RH, Massumi M, Atashi A, Izadpanah R. An efficient method for isolation of murine bone marrow mesenchymal stem cells. Int J Dev Biol. 2007; 51(8):723-729.
- NIH guide for the care and use of laboratory animals. National Institutes of Health Publication. Washington, D.C. 1985; pp. 85
- Otonkoski T, Beattie GM, Mally MI, Ricordi C, Hayek A. Nicotinamide is a potent inducer of endocrine differentiation in cultured human fetal pancreatic cells. J Clin Invest. 1993; 92(3):1459-1466.
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999; 284(5411):143-147.
- Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997; 276(5309):71-74.
- Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med. 2000; 6(3):278-282.
- Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes. 2005 ; 54(7):2060-2069.
- Sasaki M, Honmou O, Akiyama Y, Uede T, Hashi K, Kocsis JD. Transplantation of an acutely isolated bone marrow fraction repairs demyelinated adult rat spinal cord axons. Glia. 2001; 35(1):26-34.
- See EY, Toh SL, Goh JC. Multilineage potential of bone-marrow-derived mesenchymal stem cell sheets: implications for tissue engineering. Tissue Eng Part A. 2010; 16(4):1421-1431.
- Sheehan D, Hrapchak B, Theory and Practice of Histotechnology, 2nd Ed, Battelle Press, Ohil. 1980, pp: 226-227.
- Shiroi A, Yoshikawa M, Yokota H, Fukui H, Ishizaka S, Tatsumi K, Takahashi Y. Identification of insulin-producing cells derived from embryonic stem cells by zinc-chelating dithizone. Stem Cells. 2002; 20(4):284-292.
- Sjöholm A, Korsgren O, Andersson A. Polyamine requirements in nicotinamide-stimulated beta-cell differentiation in fetal porcine islet-like cell clusters. Endocrinology. 1994; 135(4):1559-1565.
- Soria B, Roche E, Berná G, León-Quinto T, Reig JA, Martín F. Insulin-secreting cells derived from embryonic stem cells normalizes glycemia in streptozotocin-induced diabetic mice. Diabetes. 2000 ;49(2):157-162.
- Südkamp NP. Clinical applications of mesenchymal stem cells. European Cells and Materials. 2007; 13 (2):3-6.
- Sun Y, Chen L, Hou XG, Hou WK, Dong JJ, Sun L, Tang KX, Wang B, Song J, Li H, Wang KX. Differentiation of bone marrow-derived mesenchymal stem cells from diabetic patients into insulin-producing cells in vitro. Chin Med J (Engl). 2007; 120(9):771-776.
- Vija L, Farge D, Gautier JF, Vexiau P, Dumitrache C, Bourgarit A, Verrecchia F, Larghero J. Mesenchymal stem cells: Stem cell therapy perspectives for type 1 diabetes. Diabetes Metab. 2009; 35(2):85-93.
- Walford RL, Gallagher R, Sjaarda JR. Serologic Typing of Human Lymphocytes with Immune Serum Obtained after Homografting. Science. 1964; 144:868-870.
- Wild S, Roglic G, Green A, Sicree R, King H. “Global prevalence of diabetes: estimates for 2000 and projections for 2030”. Diabetes Care. 2004; 27(5):1047–1053.
Table 1. Bone marrow-derived cell growth during culture
|
Total number of living cells |
|||||
|
Primary culture |
Day 0 |
Day 5 |
Day 7* |
Day 9* |
Day 14* |
|
550 ± 88.8 |
5.1 ± 0.8 |
7.77 ± 0.76 |
11.37 ± 1.01 |
15.67 ± 0.67 |
|
|
Passage one |
Day 0 |
Day 1 |
Day 2* |
Day 4* |
Day 7* |
|
1.4± 0 |
1.67 ± 0.21 |
2.93 ± 0.15 |
5.23 ± 0.31 |
8.4 ± 0.2 |
|
|
Passage two |
Day 0 |
Day 1 |
Day 2* |
Day 4* |
Day 7* |
|
1.4± 0 |
1.63 ± 0.15 |
2.3 ± 0.53 |
4.23 ± 0.31 |
7.2 ± 0.6 |
|
|
Passage three |
Day 0 |
|
|||
|
1.4± 0 |
|||||
* Significant cell growth (P < 0.05) compared to the beginning of culture with the exception of day zero of the primary culture where cultures were compared to the day 5.
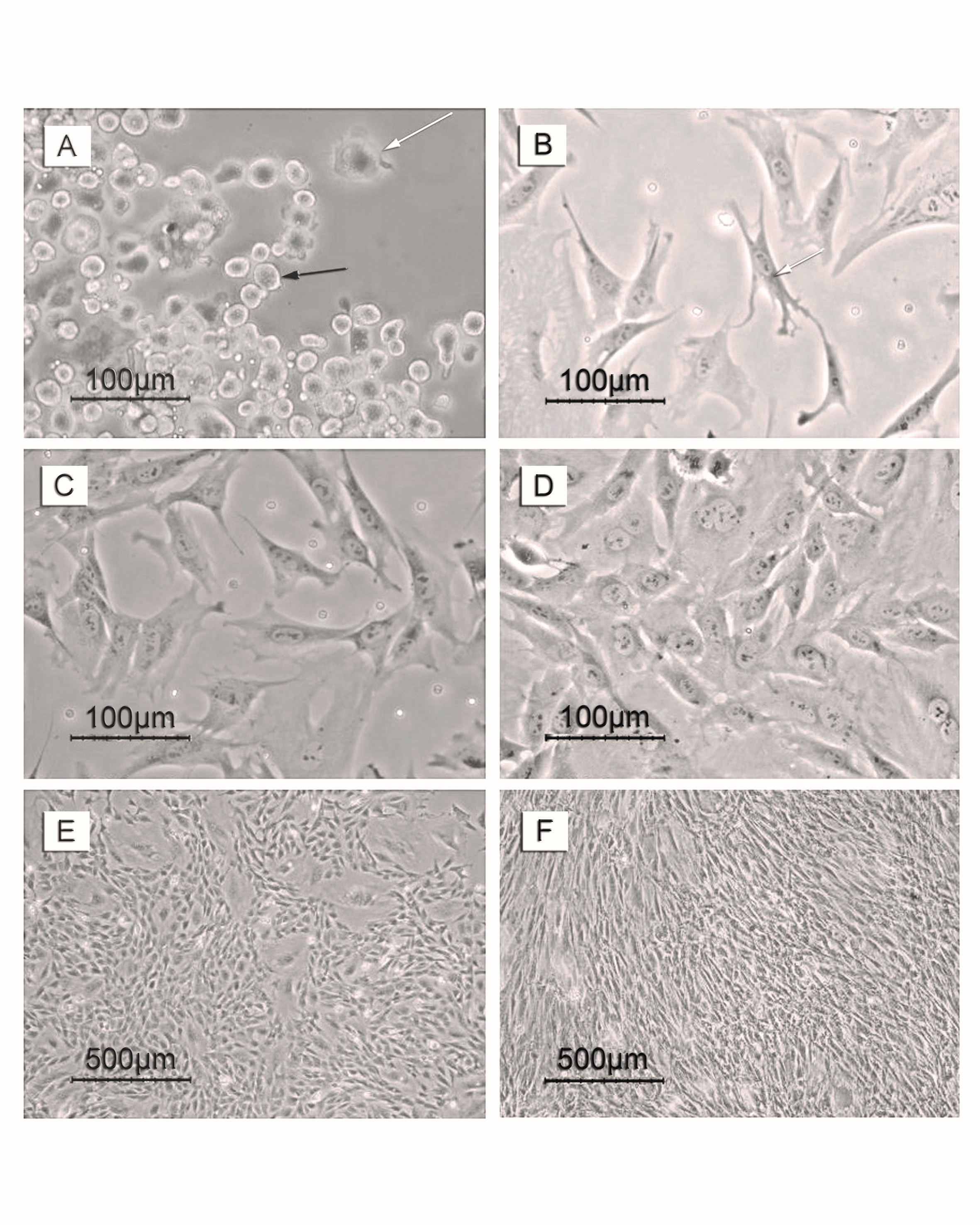
Figure 1. Photomicrographs of MSCs during the primary culture. Days of follow up included; (A) day 4, (B) day 5, (C) day 6, (D) day 7, (E) day 9 and (F) day 14. Black arrow in A refers to a red blood corpuscle and whites arrows in A and B refer to the adherent MSCs.
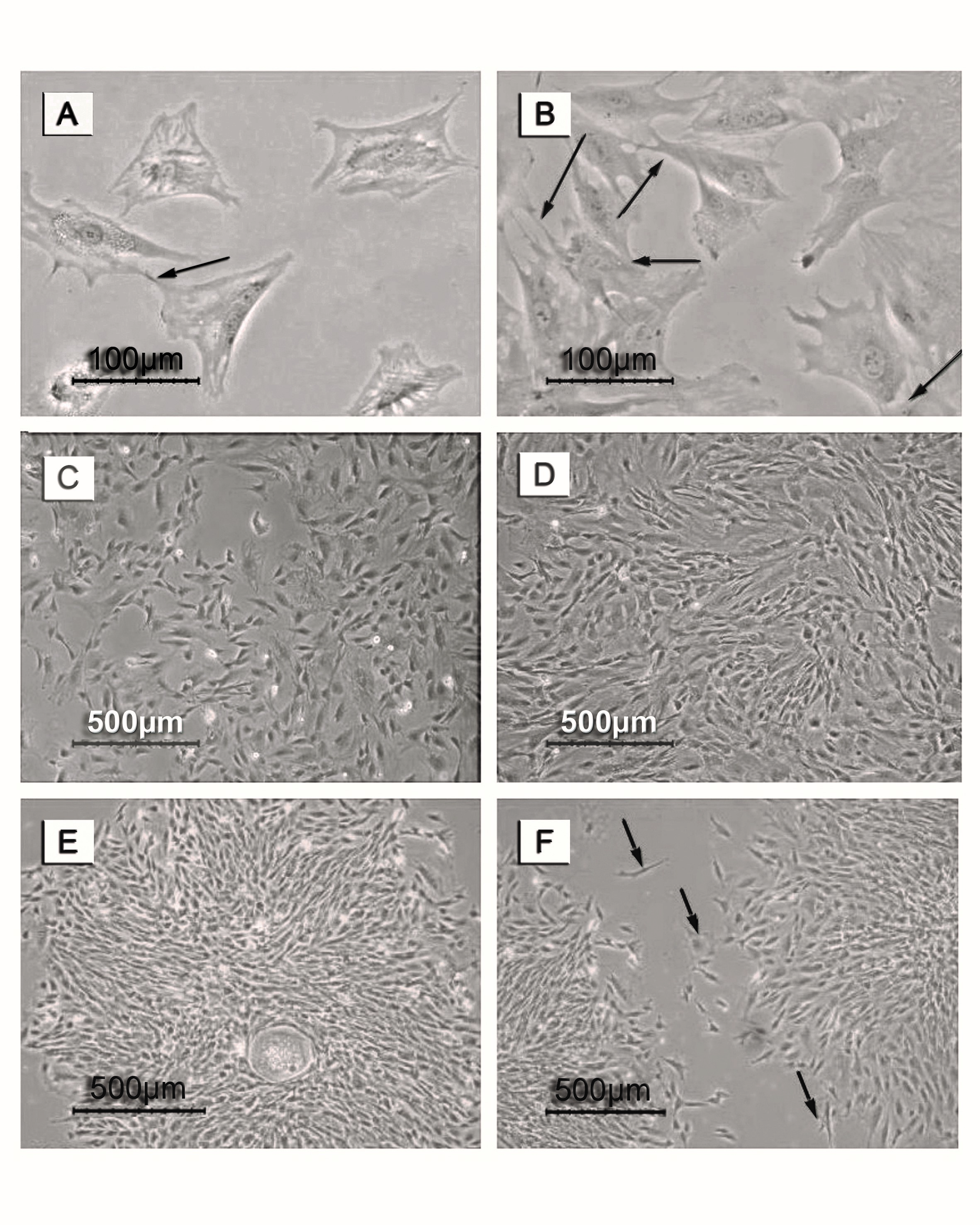
Figure 2. Photomicrographs of MSCs during passage one. Days of follow up included. (A) Day one and (B) day three, with arrows referring to cell-cell contact. (C)and (D) are representing the culture at days 4 and 6, respectively. (E) represents the morphology of the mesenchymal stem cell colony and (F) represents two proliferating colonies in their way to adhere with arrows representing cell movement.

Figure 3. Photomicrographs of MSCs at day seven during osteogenic differentiation. (A)and(B) are representing the control and tested cultures, respectively, stained with alizarin red S. (C) and (D) are representing similar control and tested cultures, respectively, stained with von Kossa. The orange to red patches in B and the brown to black patches in D indicate the osteogenic differentiation.
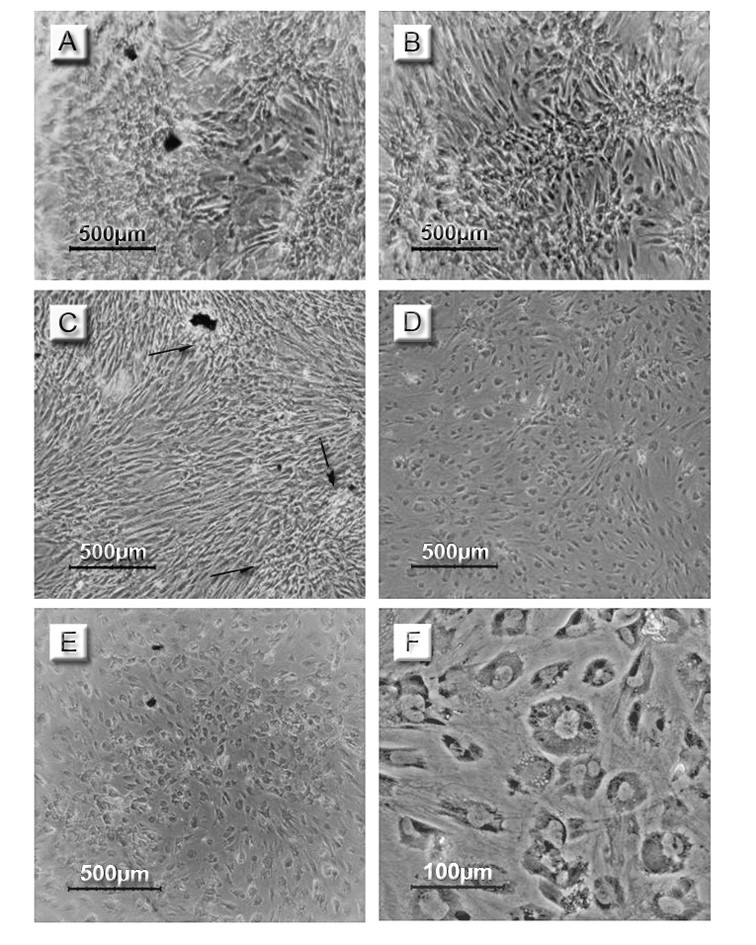
Figure 4.Photomicrographs of MSCs during the trans-differentiation into insulin-producing cells. Days of follow up included; (A, B) day 2, (C, D) day 14 and (E, F) day 19. (A, C) are representing the control cultures and (B, D, E, F) are representing the tested cultures. The control cultures showed an over confluency (arrows). The change in cell morphology of the tested cultures from the spindle-shape to the oval or rounded-shape was noted from day 14 and continued to day 19.
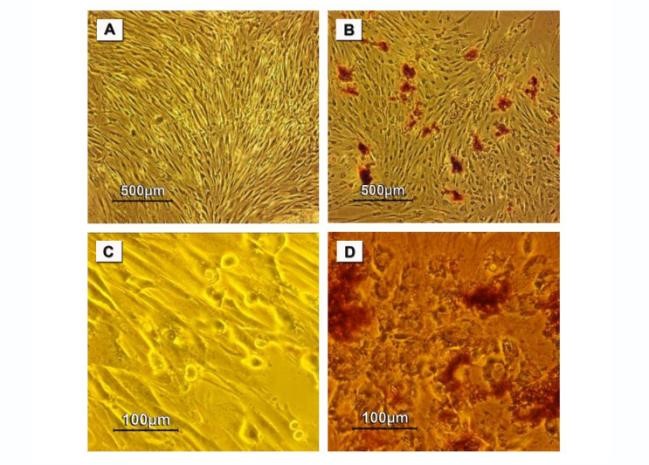
Figure 5. Photomicrographs of dithizone (DTZ) stained MSCs during the trans-differentiation into insulin-producing cells at day 14 (A, B) and day 21 (C, D). (A, C) are representing the control cultures and (B, D) are representing the tested cultures. The control cultures showed an over confluency with negative staining. While, the tested ones showed the positive staining (crimson red patches).